第4章−原子から分子ヘ:化学結合のなりたち(ムービー4-1)(パターン4-1)
一般化学の教科書は,「原子は数本の結合手によってたがいに結合し,分子を作り上げる」という記述から始まることが多い。
大学の講義では,さらに「この化学結合は硬直した棒のようなものではなくバネのような柔軟性を持ち,たえず伸縮あるいは変角振動をしている。
ただし振動があまり強く励起されると結合は切れて,組み替え(化学反応)が起こる」ことを学ぶ。
原子を結びつけて分子を構成させている「接着剤」は電子である。
この章では,最も簡単な水素分子に焦点を絞って,化学結合の形成に電子はどのように関わっているかについて概説する。(ムービー4-2)(ムービー4-3)(パターン4-2,4-3,4-4,4-5,4-6,4-7)
1.水素分子の電子状態
(1)ポテンシャル曲線(パターン4-8)
2個の水素原子をある距離![]() だけ隔てて置き,その系の位置エネルギーVを
だけ隔てて置き,その系の位置エネルギーVを![]() を横軸として図示すると,図4-1のようになる
を横軸として図示すると,図4-1のようになる
この曲線を「二原子分子のポテンシャル曲線![]() 」という。(ムービー4-4)
」という。(ムービー4-4)
この図は分子分光法により測定されたスペクトルの振動回転構造を解析することにより実験的に求めたものであるが(第7章参照),分子の波動方程式を理論的に解いても良く一致した結果が得られる。
(2.4)によれば,一般にこの曲線の勾配![]() は結合の力を表している。
は結合の力を表している。
無限遠から二つの原子が近づくにつれて結合の引力は強くなり,やがて弱くなって,![]() が最小値をとる距離
が最小値をとる距離![]() に来ると引力と反発力は釣り合って水平になる。
に来ると引力と反発力は釣り合って水平になる。
この距離![]() を「平衡距離」という。
を「平衡距離」という。
それより原子間距離を短くしようとすると強い反発力が働く。
結合がマクロの世界のバネのように伸縮振動をするのは,この引力と反発力のバランスのためである(本章2.参照)。
平衡距離でのエネルギー![]() の符号を変えた量
の符号を変えた量![]() は,各原子を
は,各原子を![]() から無限に離れた位置
から無限に離れた位置![]() まで引き離す(結合を切る)のに必要なエネルギーを表す。
まで引き離す(結合を切る)のに必要なエネルギーを表す。
Deを「(平衡)結合解離エネルギー」とよぶ。
V(re)の谷底が深いほど分子は安定で結合は強い。
水素分子では,![]() ,
,![]() である。
である。
水素分子から電子1個を除いた分子イオン![]() も安定に存在し,重要な化学種の一つである。
も安定に存在し,重要な化学種の一つである。
この系でも同様なポテンシャル曲線![]() を描くことができる。(ムービー4-5)(パターン4-9)
を描くことができる。(ムービー4-5)(パターン4-9)
平衡距離![]() と解離エネルギー
と解離エネルギー![]() は,それぞれ,
は,それぞれ,![]()
![]() である。
である。
すなわち,![]() では二つの陽子の間に強い反発力が働いているはずなのに,H2分子のおよそ60%に及ぶ
では二つの陽子の間に強い反発力が働いているはずなのに,H2分子のおよそ60%に及ぶ![]() を持つかなり強い化学結合が,わずか1個の電子によって作られている。
を持つかなり強い化学結合が,わずか1個の電子によって作られている。
(2)ボルン・オッペンハイマー近似(ムービー4-6)(パターン4-10)
水素分子は陽子2個と電子2個からなる4粒子系であり,運動の自由度(座標変数の数)は3×4 = 12個である。
このうち3個は分子の質量中心の位置,すなわち分子全体の並進運動を表す変数となる。
陽子の運動に関する他の3個の変数は,分子軸の空間的配向(すなわち分子全体の回転運動)を表す極座標θ,φ(第3章参照)と核間距離![]() (すなわち分子の振動運動)である。
(すなわち分子の振動運動)である。
残りの12 - 3 - 2 - 1=6個の変数は2個の電子がそれぞれ3次元で運動する自由度である。
陽子の質量は電子の質量に比べて3桁も大きいので,陽子の動きは電子の動きに比べると無視できるほど遅い。
したがって電子の運動を考えるとき,陽子はある位置にじっと静止しているとみなして差し支えない。
反対に陽子の運動を考えるときには電子の運動は速すぎて追随できないので,平均した形で扱えば十分である。
すなわち,電子の運動と陽子の運動は,それぞれ別々の2粒子系の方程式に分離できる(ボルン・オッペンハイマー近似)。
この近似はすべての分子で一般に良く成立することが,理論的にも実験的にも確かめられている。
以下では,まず電子の運動について説明し,そのあとで原子核(陽子)の運動(水素分子の回転と振動)について説明する。
では,電子の運動に関する波動方程式を解くときに,この近似をどのように使うのだろうか。
詳しい数式を省略して要点だけ述べると,次のようになる。
電子の運動を表すハミルトン演算子![]() と波動関数
と波動関数![]() に関して,本来は変数であるはずの核間距離
に関して,本来は変数であるはずの核間距離![]() を定数とみなし,固定された核配置に対する二つの電子の座標だけを変数とする(前章でへリウム原子の2電子系について説明したように,電子に仮に1,2と番号をつけて,それらの各3次元の座標をまとめてそれぞれ1, 2と略記する)。
を定数とみなし,固定された核配置に対する二つの電子の座標だけを変数とする(前章でへリウム原子の2電子系について説明したように,電子に仮に1,2と番号をつけて,それらの各3次元の座標をまとめてそれぞれ1, 2と略記する)。
電子の波動関数![]() をと書いて,変数1,2と定数rとを区別する。
をと書いて,変数1,2と定数rとを区別する。
また質量中心を原点として![]() に位置する二つの陽子を,それぞれa,bと表記する。
に位置する二つの陽子を,それぞれa,bと表記する。
He原子(と2電子の系)と比較すると,二つの電子の運動エネルギーを表す項は(3.48),(3.49)と同じ形であるが,クーロン力の位置エネルギー![]() は,原子核(陽子)が2個(2中心系)になったために,(3.50)と違って多数項の和になる(図4-2)。
は,原子核(陽子)が2個(2中心系)になったために,(3.50)と違って多数項の和になる(図4-2)。
第3章と同様に原子単位で表すと,電子の運動を表す波動方程式は
| (4.1) |
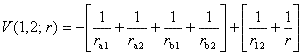 |
(4.2) |
となる。
位置エネルギーの初めの負の4項は陽子と電子のクーロン引力を表し,終わりの正の2項は電子と電子および陽子と陽子のクーロン反発力を表す。
電子のエネルギー固有値![]() は陽子間距離
は陽子間距離![]() に依存する。
に依存する。
基底状態の電子波動関数については,電子1,2の交換に対する反対称性(3.53)を考えに入れて
| (4.3) |
と表す。
![]() は,それぞれa,bの位置に原点を置く水素原子の基底状態1sの波動関数
は,それぞれa,bの位置に原点を置く水素原子の基底状態1sの波動関数 ![]() (3.35)である(一般に原子軌道(atomic orbital)略してAOとよばれる)。
(3.35)である(一般に原子軌道(atomic orbital)略してAOとよばれる)。
ここで記号を第3章でのψからχに変えた理由は,本章から分子軌道(多中心)と原子軌道(1中心)が混ざって出てくるので,それらを区別するためである。
![]() の軌道部分を
の軌道部分を![]() と
と![]() の積で表したのは,H2分子の電子波動関数が,陽子aとbのまわりでは水素原子のls電子の球対称の波動関数でほぼ近似できると想定したからである。
の積で表したのは,H2分子の電子波動関数が,陽子aとbのまわりでは水素原子のls電子の球対称の波動関数でほぼ近似できると想定したからである。
すなわち,このモデルは,「全体の波動関数は(したがって(2.31)による電子の確率分布も),二つの水素1s原子軌道の原点を核間距離![]() だけずらせて互いに独立に置いた系として近似できる」と考えたことに相当する。
だけずらせて互いに独立に置いた系として近似できる」と考えたことに相当する。
係数Nは波動関数を規格化する定数で,ここでは内容を詳しく考える必要はない。
(3) 原子価結合法
(a)ハイトラー・ロンドンの方法(ムービー4-7)(パターン4-11)
シュレーディンガーの波動力学が発表された直後(1927年)に,ハイトラーとロンドンはこの波動関数(4.3)を用いて電子エネルギー![]() の期待値(2.33)
の期待値(2.33)![]() を計算した。
を計算した。
すなわち,
| (4.4) |
に(4.1)のハミルトニアンを代入して積分を実行すると,厳密な解が数式的に求められる(![]() に含まれる項の数が多いので,(4.4)の積分はかなり複雑である)。
に含まれる項の数が多いので,(4.4)の積分はかなり複雑である)。
![]() と書いたのは,期待値は核間距離
と書いたのは,期待値は核間距離![]() により大きく変化するからである。
により大きく変化するからである。
これをの様々な値について計算し,結果をつなぎ合わせて![]() の関数としてプロットすると,図4-1とよく似た図形が得られる。
の関数としてプロットすると,図4-1とよく似た図形が得られる。
![]() と
と![]() は,それぞれ,
は,それぞれ,![]() ,
,![]() であり,実測値と比べると
であり,実測値と比べると![]() は約1.17倍に伸び,
は約1.17倍に伸び,![]() は約2/3倍と推定されている。
は約2/3倍と推定されている。
彼らの計算はこのように半定量的なものであるが,化学結合によって分子が安定化することを量子力学を用いて初めて理論的に示した重要な論文であった*1。
*1−−−−−−−−−−−−−−−−−−−−
歴史的には,ハイトラーとロンドンは初めからこの計算を意図して実行したものではなく,2個の水素原子間に働く原子間力を求めて手探りの計算をした結果,大きな結合力が偶然に得られたのだという。しかも,彼らはこの結果に戸惑い,落胆してゾンマーフェルト教授に手紙を書いたところ,この結果の重要性を教えられてそれに気づいたとのことである(ハイトラーの述懐,大野公男解説,化学の原点1,化学結合論,日本化学会編,学会出版センター(1975))。
−−−−−−−−−−−−−−−−−−−−−−−
H‐Hの化学結合がこれだけ安定化する要因はどこにあるのだろう?
この計算結果を分析したら,直感的に分かりやすい言葉で説明できるのだろうか。
この疑問に対しては,昔から多くの議論がなされてきた。
「陽子と電子の引力が陽子と陽子および電子と電子の反発力を上回るから」であることは自明のように思われるが,その内容は決して簡単ではない。
本教材のレベルでは,「パウリの原理(電子の交換に対する反対称性)に基づく波動関数(4.3)を用いたこと,すなわち量子力学的な要因が本質的であり,単に古典電磁気学を用いて電子の平均分布とクーロンの静電相互作用を考えただけでは,水素分子の安定性はまったく説明できない」と述べるにとどめる。
(b)ハイトラー・ロンドン法の改良
上記の波動関数(4.3)は,各原子a,bの位置に置かれた原子軌道に電子を1個ずつ配置して,その積の和を考えているので,結合軸の付近で電子の存在確率は増加している(図4-3)。
これは「電子対が二つの原子に共有されている」という化学者の素朴な化学結合のイメージと良く対応している。
そこで,このような波動関数を用いる計算法を原子価結合法(valence bond method),略してVB法とよぶ。(パターン4-12)
上記の近似波動関数(4.3)を改良して真の波動関数に近づけるには,変分法(第3章)を用いて最適化する。
すなわち,調節できる変分パラメーターを波動関数の中にいくつか導入して,期待値の最小値を計算する。
たとえば,(a)電子が感じている有効核電荷(第3章2-(4))のZ=1からのはずれ(ζ〜 1.19)を取り入れる。
(b)結合を作ったことによる波動関数の球対称からのひずみ(分子軸zの方向性を持つ波動関数2pz(3.27)を球対称の1s(3.19)に少し混ぜ合わせる)。
(c)二つの電子が一つの陽子のまわりに集まる構造(イオン構造H+H−)の寄与(後述)を取り入れる。
これらのパラメーターを最適化すると,期待値は実測値のおよそ86%まで近づく。(パターン4-13,4-14)
(4)分子軌道法
分子の量子化学では,上記と異なる視点に立つ分子軌道法の応用が主流になっている。(ムービー4-8)
この方法の特徴は,個々の1電子の波動関数として分子全体に広がる関数を考えることである(分子軌道(molecular orbital),略してMOとよぶ)。
全電子系の波動関数は,個々のMOの積として表す。(そのとき電子交換に対する反対称性を考慮する。)
すなわち,原子(一中心で球対称の多電子系)で有効であった1電子近似の方法(第3章3.参照)を分子(多中心で非球対称の多電子系)に拡張したものである。
H2分子の場合には,最も簡単な波動関数は(パターン4-15)
| (4.5) |
である。
![]() は,二つの陽子a,bの位置を原点とする原子軌道
は,二つの陽子a,bの位置を原点とする原子軌道![]() の和で表したものである。
の和で表したものである。
すなわち,
| (4.6) |
である。
添え字のσは,この関数が水素分子の分子軸のまわりで軸対称であることを表している(第5章1.(3)で改めて説明する)。
この分子軌道MOは各構成原子の原子軌道の和(一般には線形結合)で作られているので,Linear Combination of Atomic Orbitalsの頭文字をとってLCAOMOとよぶ。
イメージとしては,「各電子1,2が陽子aとbのまわりを等しい滞在確率で行き来している」というモデルと考えてよい。係数は規格化定数である。
この波動関数(4.5)を(4.4)に代入して期待値を計算すると,![]() と
と![]() は
は
それぞれ1.6 a0,0.098 となる。
この結果は前述のハイトラー・ロンドンの結果より少し劣るが,結合の安定性について半定量的な説明をしていることに変わりはない。
線形結合には,(4.5)のほかにもう一つの作り方がある。すなわち原子軌道の「差」をとって,
| (4.7) |
| (4.8) |
とするものである。
これを(4.4)に代入して期待値を計算すると,εは![]() の場合と符号が逆になり,
の場合と符号が逆になり,![]() の状態では二つの水素原子が近づくと不安定になり,化学結合は作られないことが分かる。
の状態では二つの水素原子が近づくと不安定になり,化学結合は作られないことが分かる。
以上の結果は電子配置図(図4-4)にまとめることができる。
この図は分子軌道法で不可欠の重要な情報である。両側のH1sの水平線は無限遠に離れた水素原子のエネルギーを表し,真中の二つのレベルは両原子が結合領域![]() まで近づいたときに作られる二つのMO,
まで近づいたときに作られる二つのMO,![]() と
と![]() のエネルギー期待値を表している。
のエネルギー期待値を表している。
![]() では核間距離
では核間距離![]() を減少させるとエネルギーが低下するので,この軌道に入った電子は結合の形成に寄与する。
を減少させるとエネルギーが低下するので,この軌道に入った電子は結合の形成に寄与する。
そこで,このような軌道を「結合性軌道(bonding orbital)とよぶ。(パターン4-16)
H2分子が安定な理由は,「2個の電子がパウリの原理に従ってスピンをα,βと対にしてこの軌道に入り,結合の形成(分子の安定化)に寄与しているためである」と解釈できる。
反対に![]() ではエネルギーがの減少とともに上昇するので,「反結合性軌道(antibonding orbital)」とよぶ(肩に*をつけたのは,反結合性という意味である)。
ではエネルギーがの減少とともに上昇するので,「反結合性軌道(antibonding orbital)」とよぶ(肩に*をつけたのは,反結合性という意味である)。
このような軌道にもし電子が入ると,分子のエネルギーはそれだけ不安定になる。
![]() (4.6)は分子の質量中心に対して左右対称になり,二つの陽子の中間領域の電子密度が大きくなっているが,
(4.6)は分子の質量中心に対して左右対称になり,二つの陽子の中間領域の電子密度が大きくなっているが,![]() (4.8)は分子の質量中心に対して左右反対称になり,中間領域の電子密度が小さくなっている(図4-4)。
(4.8)は分子の質量中心に対して左右反対称になり,中間領域の電子密度が小さくなっている(図4-4)。
このような電子密度の傾向は,これらの分子軌道が結合性あるいは反結合性になることと密接に関連している。
この電子配置図(図4-4)を使うと,水素分子イオン![]() がなぜ安定に存在するかを容易に定性的に説明できる*2。
がなぜ安定に存在するかを容易に定性的に説明できる*2。
すなわち,1個の電子が結合性軌道![]() に入るからである。
に入るからである。
![]() ではH2と違って電子間の反発力がないので,結合性軌道のエネルギーはH2の場合に比べて少し低くなる。
ではH2と違って電子間の反発力がないので,結合性軌道のエネルギーはH2の場合に比べて少し低くなる。
(1)で述べたようにのはH2の![]() は、H2の
は、H2の![]() の約59%で,半分を少し超えているのはこのためである。
の約59%で,半分を少し超えているのはこのためである。
*2−−−−−−−−−−−−−−−−−−−−
この電子配置図をさらに応用すると,He2の系はなぜ安定に存在できないかも説明できる。He2は4電子系である。二つのHe原子を接近させると,上記の水素分子の場合と同様に二つのls原子軌道の線形結合によって結合性と反結合性の分子軌道が作られる。そこに4個の電子を入れると,電子の結合エネルギーは両軌道で打ち消されて,ポテンシャル曲線![]() には有意の極小値が現れないことを定性的に理解できる。最近の実験結果によれば,平衡位置He2の
には有意の極小値が現れないことを定性的に理解できる。最近の実験結果によれば,平衡位置He2の![]() は
は![]() で,平衡結合解離エネルギー
で,平衡結合解離エネルギー![]() はわずかに
はわずかに![]() である。
である。
−−−−−−−−−−−−−−−−−−−−−−−−
(5)分子軌道法の変分法
最も簡単な分子軌道の波動関数(4.5,6)を用いて求めたエネルギー期待値は,原子価結合法による期待値に比べて不満足なものであった。
その原因は,この波動関数を仮定するとイオン構造H+H−の寄与を過大評価してしまうことにある。
すなわち,(4.6)を(4.5)に代入して書き換えると
| (4.9) |
となり,右端の電子スピンと規格化の因子を省略して書くと
| (4.10) |
となる。
初めの二つの項はVB法の波動関数(4.3)と同一で,電子はそれぞれ異なる陽子のまわりに存在することを想定しているが,それに続く二つの項は2電子がaまたはbの陽子のまわりに存在することを表しているので,イオン構造H+H−が1:1の割合で混ざりあった状態を想定していることになる。
分子軌道法では,(4.5,6)のように各電子がまったく独立に陽子aとbの付近に存在できると考えているので,二つの電子がたまたま同一の陽子の付近に存在する確率を過大評価してしまう。
化学的な常識では,電子がこれほどひどく偏在するとは考えられない。
原子価結合法では,イオン構造の混ざり方を表すパラメーター![]() を入れて試行関数を次式のように作り,
を入れて試行関数を次式のように作り,
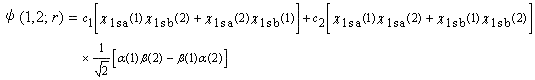 |
(4.11) |
変分法で実際に最適化してみると,![]() はおよそ0.175となり,(4.10)で想定した0.50よりはるかに小さくなる。
はおよそ0.175となり,(4.10)で想定した0.50よりはるかに小さくなる。
電子の存在確率は波動関数の2乗で表されるので(第2章の仮定3),イオン構造の割合は,およそ![]() となる。
となる。
変分法を用いれば,分子軌道法でもこのイオン構造の寄与の大きさを正しく評価することができる。
そのためには,励起状態の電子配置![]() に対応する波動関数を基底状態の電子配置
に対応する波動関数を基底状態の電子配置![]() に対応する波動関数に少し混ぜて,その係数
に対応する波動関数に少し混ぜて,その係数![]() を最適化すればよい。
を最適化すればよい。
すなわち,
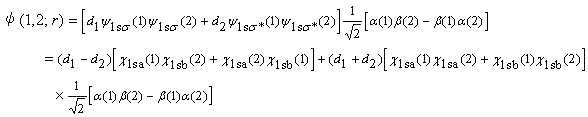 |
(4.12) |
となるので,![]() が(4.11)の
が(4.11)の![]() に相当する。
に相当する。
さらに有効核電荷およびls原子軌道に対する2p原子軌道の混ぜ合わせを考慮して最適化すると,(4.11)と同様の結果が得られる。
このように,異なる電子配置に対応する波動関数を混ぜて最適化する方法を配置間相互作用(configuration interaction)とよび,分子軌道法の重要な方法の一つとなっている。(ムービー4-9)(パターン4-17)
へリウム原子の場合と同様に,水素分子のスペクトルを解析すると,電子励起状態について詳細な知見が得られる(図4-6)。
ポテンシャル曲線が極小値を持ち,たとえ弱くても何らかの結合が存在する場合のほかに,曲線が単調に右下がりになる状態も多い。
後者の状態は,生成すると二つの水素原子間に反発力が働いて,すぐに解離してしまう。
二つの電子の分子軌道への入り方には,二つが等しい分子軌道を占める場合とそれぞれ異なる分子軌道を占める場合とがある。
前者ではパウリの排他原理により一重項だけが許され,後者では一重項のほかに三重項状態も現れる(第3章2.6参照)。